COMPUTATIONAL GENOMICS FOR DRUG ABUSE (CG4DA)
PI: Dr. Eric Xing, Ph.D and Co-PI: Wei Wu
Current Research
The objectives of Core C, Computational Genomics Core for DA (CG4DA), are to help support the FRPs and to address fundamental methodological challenges of unraveling the genetic basis of DA and medication research by a systematic inference of the mapping between genetic variations and susceptibility to DA possibly induced by certain chemical compounds. Such a mapping provides a genome-wide atlas of potential targets and their risk under chemical compounds. The three themes of the services that CG4DA proposed to provide to the FRPs and also a broader DAR community are: (i) developing machine learning methods for transcriptome-wide screening of expression traits and molecular markers for DA; (ii) genome-wide discovery of new drug targets and their epistatic genetic influences via structured association mapping; and (iii) software development for DA diagnosis, and towards guiding DA treatment.
Genome-Wide Association Studies of Dynamic Complex Traits (Marchetti-Bowick, M., et. al., 2016)
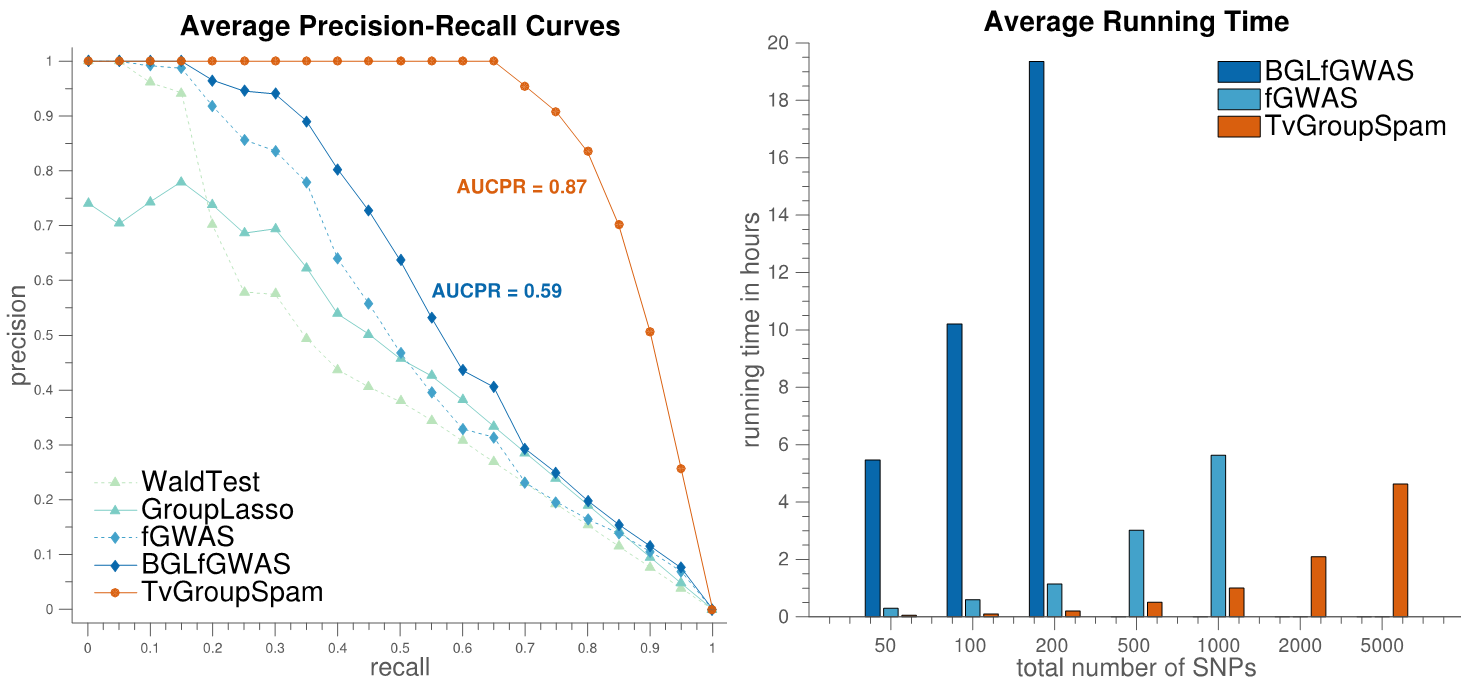
Despite the widespread popularity of genome-wide association studies (GWAS) for genetic mapping of complex traits, most existing GWAS methodologies are still limited to the use of static phenotypes measured at a single time point. In this work, we propose a new method for association mapping that considers dynamic phenotypes measured at a sequence of time points. Our approach relies on the use of Time-Varying Group Sparse Additive Models (TV-GroupSpAM) for high-dimensional, functional regression. This new model detects a sparse set of genomic loci that are associated with trait dynamics, and demonstrates increased statistical power over existing methods. Our model has three major advantages over existing approaches: (i) we leverage dynamic trait data; (ii) we model the contribution of each SNP to the phenotype as a smooth function of time, and explicitly learn these influence patterns; (iii) we model the combined effects of multiple SNPs on the phenotype and select a sparse subset that participate in the model, thereby identifying meaningful SNP-trait associations. Our results show that TV-GroupSpAM exhibits desirable empirical advantages over baseline methods on both simulated and real datasets.
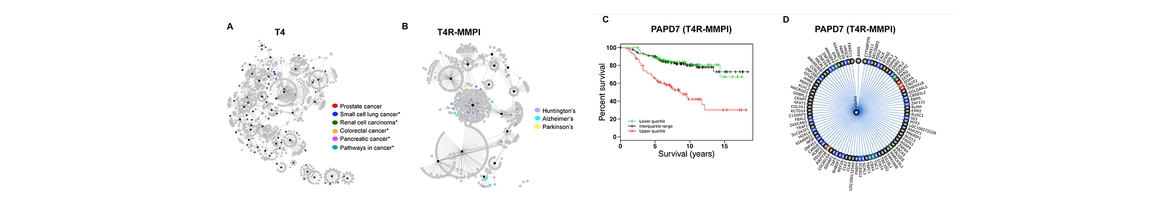
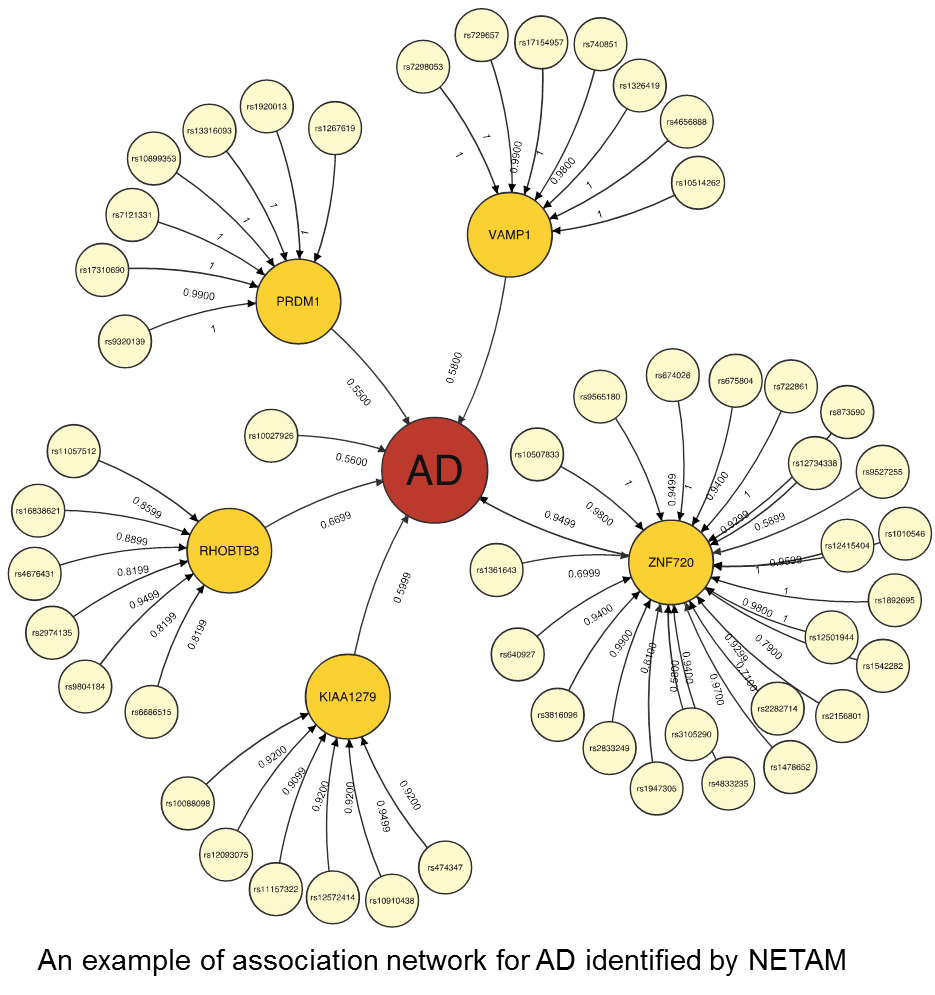
A network-driven approach for genome-wide association mapping (Lee S, et. al., 2016)
It remains a challenge to detect associations between genotypes and phenotypes because of insufficient sample sizes and complex underlying mechanisms involved in associations. Fortunately, it is becoming more feasible to obtain gene expression data in addition to genotypes and phenotypes, giving us new opportunities to detect true genotype-phenotype associations while unveiling their association mechanisms. In this work, we develop a novel method, NETAM, that accurately detects associations between SNPs and phenotypes, as well as gene traits involved in such associations. We take a network-driven approach: NETAM first constructs an association network, where nodes represent SNPs, gene traits or phenotypes, and edges represent the strength of association between two nodes. NETAM assigns a score to each path from an SNP to a phenotype, and then identifies significant paths based on the scores. In our simulation study, we show that NETAM finds significantly more phenotype-associated SNPs than traditional genotype-phenotype association analysis under false positive control, taking advantage of gene expression data. Furthermore, we applied NETAM on late-onset Alzheimer’s disease data and identified 477 significant path associations, among which we analyzed paths related to beta-amyloid, estrogen, and nicotine pathways. We also provide hypothetical biological pathways to explain our findings.
Genome-wide Detection of Marginal and Interacting Genetic Variations (Lee, S, et. al., 2016)
Genome-wide association studies have revealed individual genetic variants associated with phenotypic traits such as disease risk and gene expressions. However, detecting pairwise interaction effects of genetic variants on traits still remains a challenge due to a large number of combinations of variants (∼1011 SNP pairs in the human genome), and relatively small sample sizes (typically <104). Despite recent breakthroughs in detecting interaction effects, there are still several open problems, including: (1) how to quickly process a large number of SNP pairs, (2) how to distinguish between true signals and SNPs/SNP pairs merely correlated with true signals, (3) how to detect nonlinear associations between SNP pairs and traits given small sample sizes, and (4) how to control false positives. In this work, we develop a unified framework, called SPHINX, which addresses the aforementioned challenges. We first propose a piecewise linear model for interaction detection, because it is simple enough to estimate model parameters given small sample sizes but complex enough to capture nonlinear interaction effects. Then, based on the piecewise linear model, we introduce randomized group lasso under stability selection, and a screening algorithm to address the statistical and computational challenges mentioned above. Our results demonstrate that SPHINX achieves better power than existing methods for interaction detection under false positive control. We further applied SPHINX to late-onset Alzheimer’s disease dataset, and report 16 SNPs and 17 SNP pairs associated with gene traits. We also present a highly scalable implementation of our screening algorithm, which can screen ∼118 billion candidates of associations on a 60-node cluster in <5.5 hours.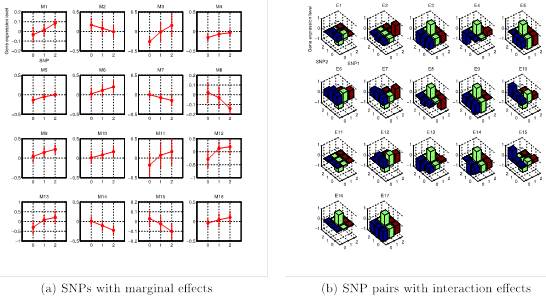
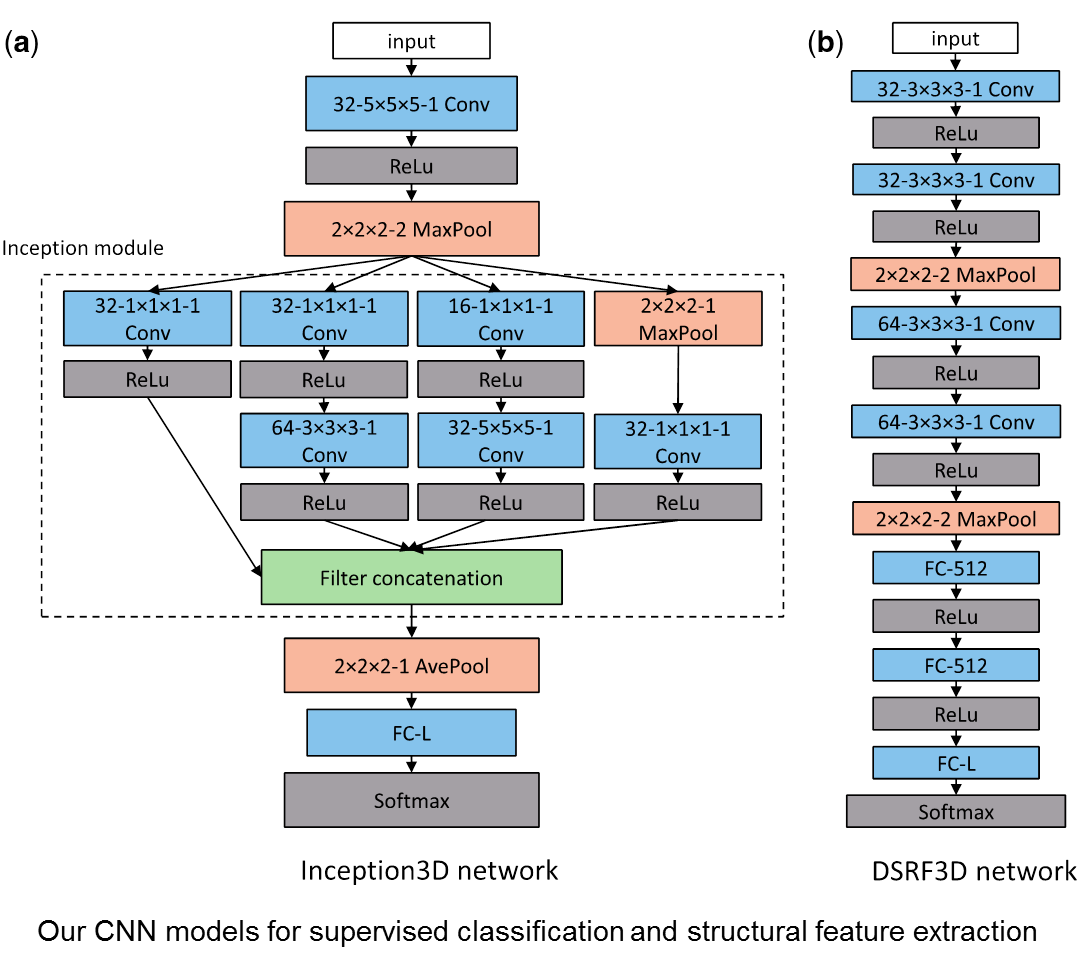
Deep learning-based subdivision approach for large scale macromolecules structure recovery from electron cryo tomograms (Xu, M., et. al., 2017)
Cellular Electron CryoTomography (CECT) enables 3D visualization of cellular organization at near-native state and in sub-molecular resolution, making it a powerful tool for analyzing structures of macromolecular complexes and their spatial organizations inside single cells. However, high degree of structural complexity together with practical imaging limitations makes the systematic de novo discovery of structures within cells challenging. It would likely require averaging and classifying millions of subtomograms potentially containing hundreds of highly heterogeneous structural classes. Although it is no longer difficult to acquire CECT data containing such amount of subtomograms due to advances in data acquisition automation, existing computational approaches have very limited scalability or discrimination ability, making them incapable of processing such amount of data. In this work, we develop a new approach for subdividing subtomograms into smaller but relatively homogeneous subsets. The structures in these subsets can then be separately recovered using existing computation intensive methods. Our approach is based on supervised structural feature extraction using deep learning, in combination with unsupervised clustering and reference-free classification. Our experiments show that, compared with existing unsupervised rotation invariant feature and pose-normalization based approaches, our new approach achieves significant improvements in both discrimination ability and scalability. More importantly, our new approach is able to discover new structural classes and recover structures that do not exist in training data.
Software, methods and reference
GenAMap
– Curtis, Ross E., Peter Kinnaird, and Eric P. Xing. “GenAMap: visualization strategies for structured association mapping.” Biological Data Visualization (BioVis), 2011 IEEE Symposium on. IEEE, 2011.
– Xing, Eric P., et al. “GWAS in a box: statistical and visual analytics of structured associations via GenAMap.” PloS one 9.6 (2014): e97524.
– Wang, Haohan,et al. “GenAMap on Web: Visual Machine Learning for Next Generation GWAS” in preparation.
Confounder filtering
– Wang, Haohan, Zhenglin Wu, and Eric P. Xing. “Removing Confounding Factors Associated Weights in Deep Neural Networks Improves the Prediction Accuracy for Healthcare Applications.” PSB. 2019.
CMM
– Wang, H., Liu, M., Lee, S., Vanyukov, M. M., Wu, W., & Xing, E. P. (2018). Coupled Mixed Model for joint genetic analysis of complex disorders from independently collected data sets: application to Alzheimer’s disease and substance use disorder. BioRxiv, 336727.
Precision Lasso
– Wang Haohan, Benjamin J. Lengerich, Bryon Aragam and Eric P. Xing, “Precision Lasso: Accounting for Correlations and Linear Dependencies in High-Dimensional Genomic Data” In revision
CS-LMM / LRVA
– Wang, Haohan, et al., “Discovering Weaker Genetic Associations with Validated Association, with Studies of Alzheimer’s Disease and Drug Abuse Disorder”
MKKC
– Bang, Seojin, Yaoliang Yu, and Wei Wu. 2019. “Robust Multiple Kernel K-Means Clustering Using Min-Max Optimization.” ArXiv Preprint ArXiv:1803.02458.